
The user-friendly and Galaxy-supported pipeline FROGS analyses large sets of DNA amplicons sequences accurately and rapidly, essential for microbe community studies.
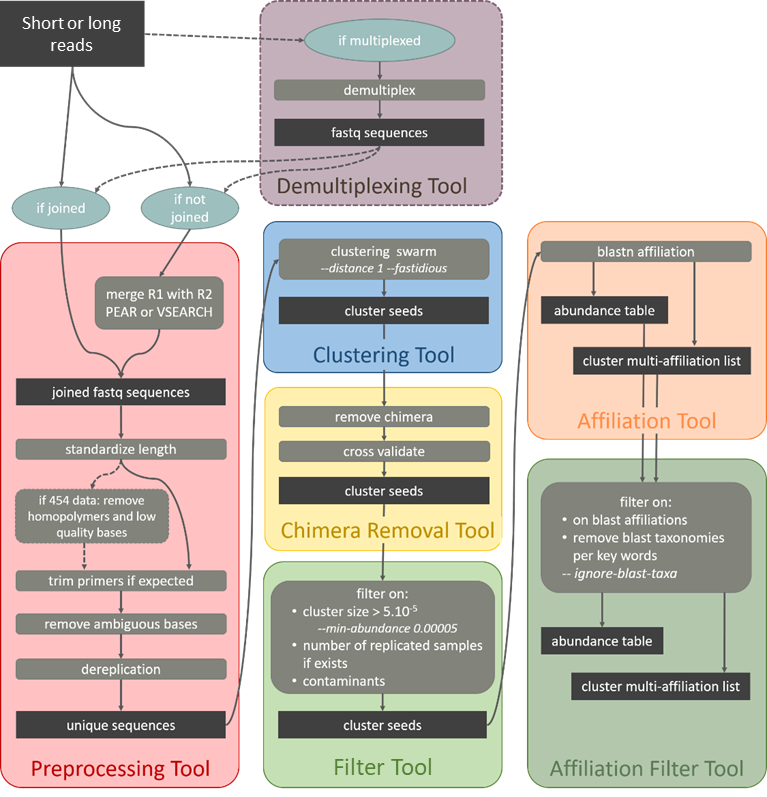
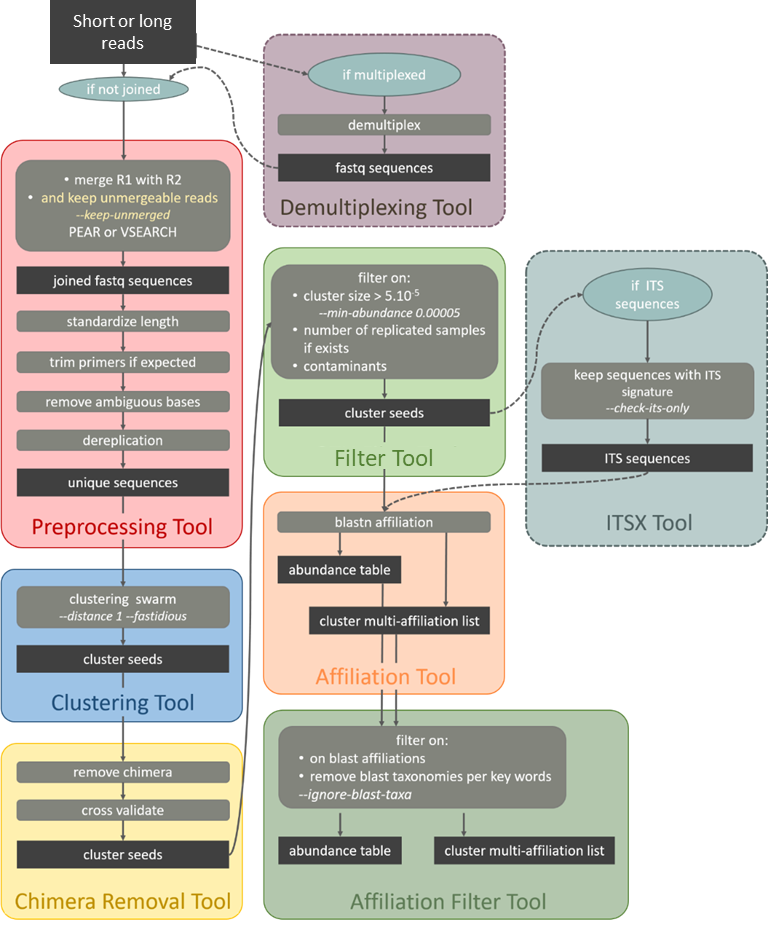
- FROGS was designed to support multiplexed and demultiplexed sequences.
- FROGS supports 16S, 18S, 23S, ITS and others amplicon reads.
- FROGS supports short or long reads.
- The preprocessing tool is dedicated to paired sequences merging, cleaning and dereplication.
- The clustering tool uses Swarm with a local clustering threshold, not a global clustering threshold like other software do.
- Chimera removal tool uses VSEARCH combined with an innovative chimera cross-validation.
- A filtering tool allows to remove noisy data.
- Affiliation tool returns taxonomic affiliation for each ASV using two methods with a unique multi-affiliation output.
- FROGS offers numerous banks for affiliation step (cf. list ).
- A lot of statistical results and numerous graphical illustrations are also produced.
- FROGS is designed for non-specialists thanks to its Galaxy interface, but is also available with command lines: github.
- Its tools can be used independently, or as a workflow.
- To install FROGS: conda or galaxy toolshed.
FROGS was tested on many datasets
- The "FROGS 16S Benchmark tab" and "FROGS ITS Benchmark tab" show comparisons between FROGS and other popular pipelines.
Standard Operation Procedure for amplicons
i.e. 16S, rpob, coi, 18S, ...
i.e. 16S, rpob, coi, 18S, ...
Standard Operation Procedure for data with unmergeable amplicons
i.e. ITS, rpb2, d1d2, ...
i.e. ITS, rpb2, d1d2, ...
Citation
The publications :
Frédéric Escudié, Lucas Auer, Maria Bernard, Mahendra Mariadassou, Laurent Cauquil, Katia Vidal, Sarah Maman, Guillermina Hernandez-Raquet, Sylvie Combes, Géraldine Pascal; FROGS: Find, Rapidly, OTUs with Galaxy Solution, Bioinformatics, Volume 34, Issue 8, 15 April 2018, Pages 1287–1294
Maria Bernard, Olivier Rué, Mahendra Mariadassou and Géraldine Pascal; FROGS: a powerful tool to analyse the diversity of fungi with special management of internal transcribed spacers, Briefings in Bioinformatics 2021, 10.1093/bib/bbab318
Frédéric Escudié, Lucas Auer, Maria Bernard, Mahendra Mariadassou, Laurent Cauquil, Katia Vidal, Sarah Maman, Guillermina Hernandez-Raquet, Sylvie Combes, Géraldine Pascal; FROGS: Find, Rapidly, OTUs with Galaxy Solution, Bioinformatics, Volume 34, Issue 8, 15 April 2018, Pages 1287–1294
Maria Bernard, Olivier Rué, Mahendra Mariadassou and Géraldine Pascal; FROGS: a powerful tool to analyse the diversity of fungi with special management of internal transcribed spacers, Briefings in Bioinformatics 2021, 10.1093/bib/bbab318
To test FROGS
Install FROGS: conda or galaxy toolshed.
FROGS training documentations and training data : Formation data and documentation
Redo FROGS'assessments with command lines for analyse 16S amplicons Get data built with sequences from utax and silva databanks: doi.org/10.15454/VGVCIJ
Redo FROGS'assessments with command lines for analyse ITS amplicons Get data built with sequences from UNITES: doi.org/10.15454/AOT7UL
FROGS training documentations and training data : Formation data and documentation
Redo FROGS'assessments with command lines for analyse 16S amplicons Get data built with sequences from utax and silva databanks: doi.org/10.15454/VGVCIJ
Redo FROGS'assessments with command lines for analyse ITS amplicons Get data built with sequences from UNITES: doi.org/10.15454/AOT7UL
License
GNU GPLv3 (details)
Legal terms






